DeltaMass: view and interrogate open search proteomics data

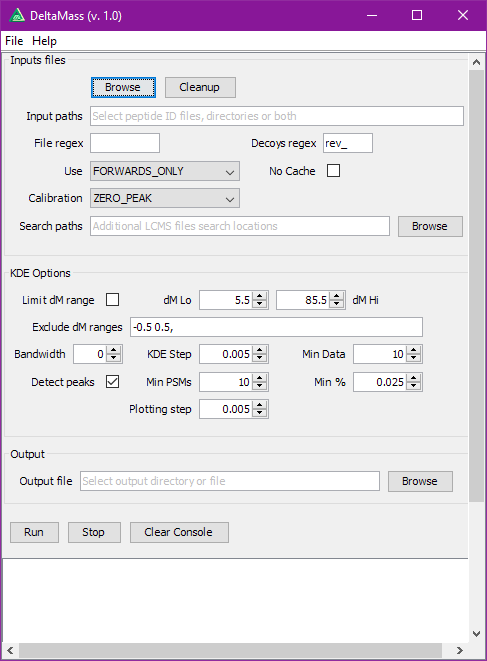
DeltaMass will plot mass-differences from a proteomic “Open Search”. An “Open Search” is any search of fragmentation spectra (MS2) against a protein database when the precursor mass tolerance was set to a high enough value. E.g. not the usual 5-10-20 ppm, but instead 50-100-200 Da. The result of such a search is quite different from the regular one, it might contain a lot of spurious hits, but a lot of hits will still correspond to reasonable matches.
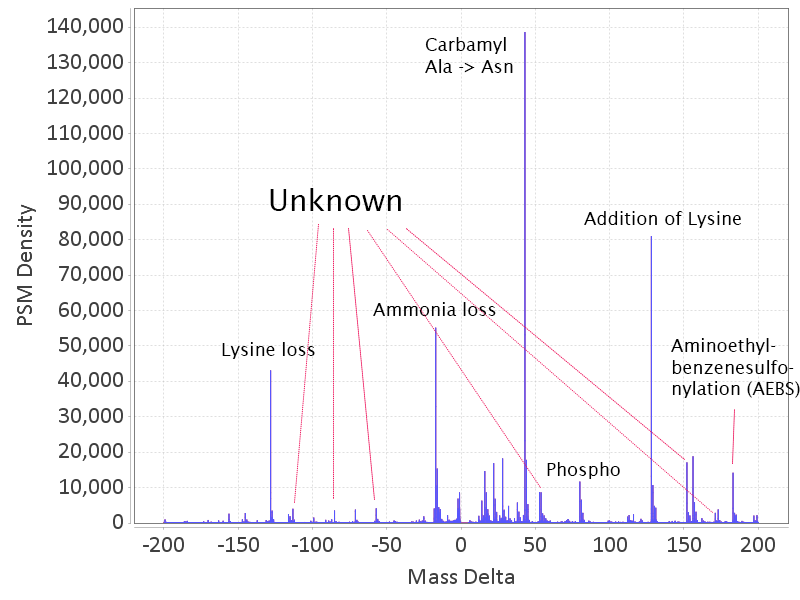
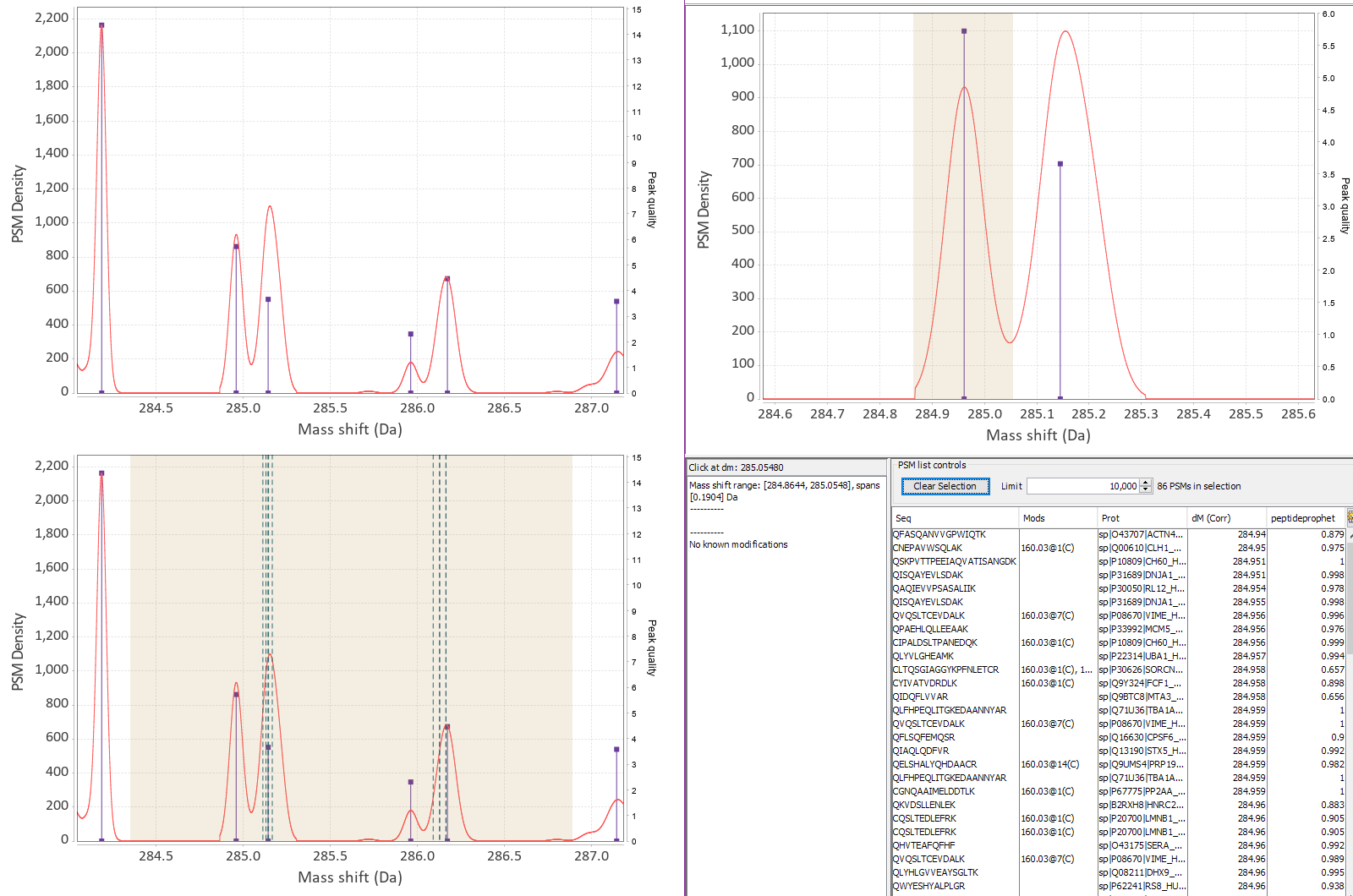
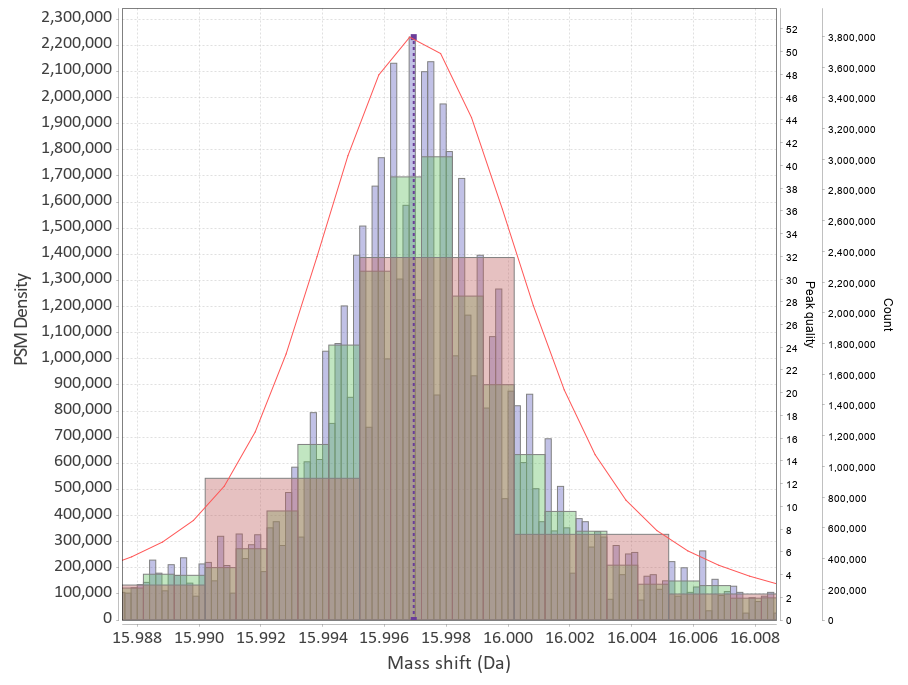
The differences in mass between the observed value for a precursor and the theoretical peptide mass from a database might be many Daltons. Nevertheless, you’ll still see lots of similar mass differences, which should correspond to Post Translational Modifications (PTMs) or chemical modifications or other artifacts in the data.
After performing an Open Search you might want to identify the differences to maybe include them into the variable modifications list of your search engine settings. You might also discover unexpected artifacts, like chemical derivatives of peptides occurring because of your sample preparation protocols etc.
This is where this program comes into play.
Referencing work / Citing
If you have used this software in your work or research, please cite:
| Manuscript | Dmitry M. Avtonomov, Andy Kong, and Alexey I. Nesvizhskii, "DeltaMass: Automated Detection and Visualization of Mass Shifts in Proteomic Open-Search Results". J. Proteome Res., 2019, 18 (2), pp 715–720 |
| DOI | 10.1021/acs.jproteome.8b00728 |
| BibTex |
@article{Avtonomov2019,
author = {Avtonomov, Dmitry M. and Kong, Andy and Nesvizhskii, Alexey I.},
title = {DeltaMass: Automated Detection and Visualization of Mass Shifts in Proteomic Open-Search Results},
journal = {J. Proteome Res.},
publisher = {American Chemical Society},
year = {2019},
volume = {18},
number = {2},
pages = {715--720},
url = {https://doi.org/10.1021/acs.jproteome.8b00728},
doi = {https://doi.org/10.1021/acs.jproteome.8b00728}
}
|